The occurrence of SSU tRF polygons related to those from either Azospirillum brasilense, Escherichia coli or Rhizobium leguminosarum.
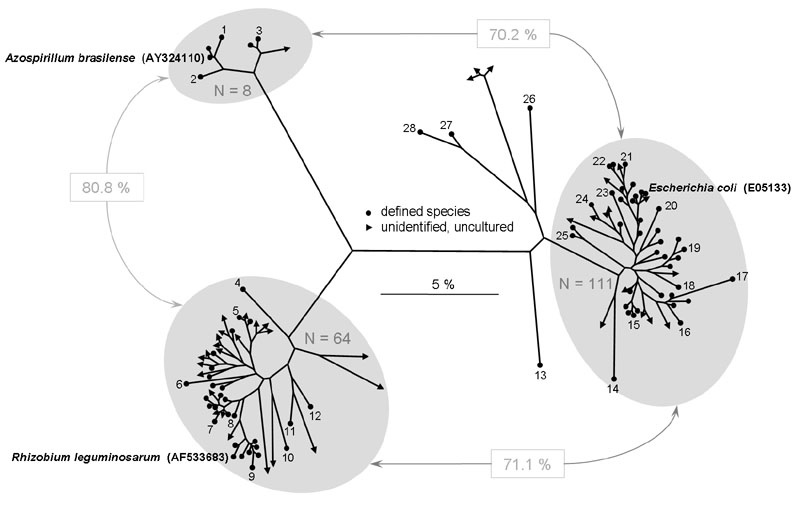
The TReFID program provided 192 polygons related to those of the three organisms and deposited in the TReFID databank (8 for A. brasilense, 111 for E. coli and 64 for R. leguminosarum). Their sequences were used to construct the phylogenetic tree using the Neighbor-Joining method. The tree shows that only six artifacts were obtained (see text). The average pairwise sequence homology between the three clusters of organisms was 70.2, 80.8 and 71.1 %, respectively.
The numbers in the figure refer to:
| Strain | GenBank link | Strain | GenBank link | |||
| 1 | Roseomonas fauriae | AF533354 | 15 | Pectobacterium carotovorum | AF373189 | |
| 2 | Roseomonas genomospecies | AY150050 | 16 | Serratia quinovorans | AJ279050 | |
| 3 | Azospirillum sp. | AB049110 | 17 | Serratia sp. | AF511524 | |
| 4 | Devosia riboflavina | AF501346 | 18 | Serratia odorifera | AF286870 | |
| 5 | Agrobacterium tumefaciens | AF508094 | 19 | Pectobacterium carotovorum | AF373184 | |
| 6 | Agrobacterium tumefaciens | AF406666 | 20 | Erwinia amylovora | AF141892 | |
| 7 | Sinorhizobium kummerowiae | AF364067 | 21 | Escherichia albertii | AJ508775 | |
| 8 | Sinorhizobium meliloti | AF533685 | 22 | Escherichia coli | AY319394 | |
| 9 | Rhizobium tropici | U89832 | 23 | Obesumbacterium proteus | AY077753 | |
| 10 | Mesorhizobium plurifarium | AF516882 | 24 | Escherichia senegalensis | AY217654 | |
| 11 | Ochrobactrum sp. | AF452128 | 25 | Dickeya dadantii | AF520707 | |
| 12 | Ochrobactrum anthropi | AF501340 | 26 | Shewanella gelidimarina | AF530149 | |
| 13 | Idiomarina baltica | AJ440214 | 27 | Pseudoalteromonas sp. | AB055788 | |
| 14 | Xenorhabdus nematophila | AF522294 | 28 | Pseudoalteromonas prydzensis | U85855 |
Detailed Explanations:
To test the “phylogenetic separation” of the TReFID method, theoretically derived tRF values for three proteobacterial strains were used as input data instead of experimentally obtained tRFs. These three strains were selected from the TReFID SSU database (version A), but the original data were taken from GenBank (accession numbers in parentheses):
Azospirillum brasilense
Sp7 (AY324110)
Escherichia coli K12 (E05133)
Rhizobium leguminosarum
WSM1698 (AF533683)
The in silico analysis was based on these strains' calculated tRF values for thirteen restriction enzymes: AluI (cg/at), Bme1390I (cc/Ngg), Bsh1236I (cg/cg), Cfr13I (g/gNcc), HaeIII (gg/cc), Hin6I (c/gcg), HinfI (g/aNtc), MboI (/gatc), MspI (c/cgg), RsaI (gt/ac), TaiI (acgt/), TaqI (t/cga), and TasI (/aatt). However, none of the three strains provided tRFs for all thirteen enzymes; in some cases there was no restriction site in the 500 nt downstream of the 63f primer binding motif. For instance, RsaI provided a tRF for E. coli but not for A. brasilense or R. leguminosarum as those two sequences had restriction sites only beyond the 500 nt border. The calculated tRF values as stored in the TReFID database are given in the table below:
|
|
A. brasilense |
E. coli |
R. leguminosarum |
|
AluI |
226 |
40 |
171 |
|
Bme1390I |
415 |
(out of range) |
402 |
|
Bsh1236I |
173 |
360 |
60 |
|
Cfr13I |
167 |
161 |
255 |
|
HaeIII |
36 |
171 |
190 |
|
Hin6I |
45 |
336 |
302 |
|
HinfI |
274 |
295 |
66 |
|
MboI |
217 |
238 |
204 |
|
MspI |
115 |
461 |
366 |
|
RsaI |
(out of range) |
392 |
(out of range) |
|
TaiI |
65 |
143 |
481 |
|
TaqI |
(out of range) |
(out of range) |
489 |
|
TasI |
470 |
(out of range) |
457 |
(For the original data refer to this page or the project file.)
These tRF values were analysed with the SSU databases (both version A and B) using the default parameters (the TReFID output files for both databases can be downloaded here):
(1) The error interval in fragment sizing was set to plus/minus 0.50 %, meaning a tRF of 200 nt was treated not as a discrete value but as an interval [199;201]. So, any database sequence providing a tRF of 199, 200, or 201 nt would get a score of 1.00 for the respective restriction enzyme. A score of 0.50 would be allocated for each match within 200 plus/minus 1.00 % = [198;202], and a score of 0.25 for each match within 200 plus/minus 1.50 % = [197;203].
(2) The number of restriction enzymes with matches had to be at least two thirds the number of enzymes providing a tRF for a given database sequence. For example, a sequence providing tRFs for all thirteen restriction enzymes employed had to match at least in nine of these enzymes with tRFs from the input data to be included in the results list.
The relevance of these ppoints is illustrated by the following example of two sequence - one from the results list (Escherichia sp.) and one which was discarded during the analysis (Neisseria meningitidis). In case of Escherichia three enzymes provided no tRFs in the sequence region examined. Concerning the ten remaining enzymes one (Cfr13I) yielded no match with the input data: a 170 nt tRF was expected for this Escherichia sequence, but the tRFs from the input were 161, 167, and 255 nt in length and thus did not match. Seven other enzymes did match exactly and therefore gave maximal scores of 1.00. In two cases, low-scoring matches occured: score 0.50 for HaeIII as the found tRF (171 nt) differed by more than 0.50 % but less than 1.00 % from the expected one (170 nt) and score 0.25 for TaiI because the deviation of found and expected tRFs was between 1.00 % and 1.50 %. For a complete list of the TReFID results for Control C see here.
|
Escherichia sp. (X80733) |
Neisseria meningitidis (AF310423) |
||||||
| Enzyme | Expected | Found | Score | Expected | Found | Score | |
| AluI: | 40 | 40.00 | 1.00 | 40 | 40.00 | 1.00 | |
| Bme1390I: | 572 | (out of range) | 0.00 | 571 | (out of range) | 0.00 | |
| Bsh1236I: | 361 | 360.00 | 1.00 | 360 | 360.00 | 1.00 | |
| Cfr13I: | 170 | (no match) | 0.00 | 169 | 167.00 | 0.25 | |
| HaeIII: | 172 | 171.00 | 0.50 | 171 | 171.00 | 1.00 | |
| Hin6I: | 337 | 336.00 | 1.00 | 176 | (no match) | 0.00 | |
| HinfI: | 296 | 295.00 | 1.00 | 295 | 295.00 | 1.00 | |
| MboI: | 239 | 238.00 | 1.00 | 112 | (no match) | 0.00 | |
| MspI: | 462 | 461.00 | 1.00 | 461 | 461.00 | 1.00 | |
| RsaI: | 393 | 392.00 | 1.00 | 91 | (no match) | 0.00 | |
| TaiI: | 145 | 143.00 | 0.25 | 91 | (no match) | 0.00 | |
| TaqI: | 781 | (out of range) | 0.00 | 115 | (no match) | 0.00 | |
| TasI: | 517 | (out of range) | 0.00 | 320 | (no match) | 0.00 | |
| Score sum: | 7.75 of 10.00 | 5.25 of 12.00 | ||
| Number of matches: | 9 of 10 | 6 of 12 | ||
| >> conditions met | >> conditions not met |
The TReFID analysis resulted in 537 hits, i.e. 537 partial SSU sequences out of 17,462 contained in the database used (version A, see below). These sequences were then subjected to a phylogenetic analysis. All sequences of less than 500 nt in length were deleted prior to a multiple sequence alignment (ClustalX). As a result, only 257 of 537 sequences remained in the list. Most of the 280 short sequences deleted belonged to Salmonella enterica (209 cases); the rest were sequences from the genera Escherichia and Rhizobium and of many uncultured bacteria without lineage annotations. After the provisory alignment nine additional sequences with less than 600 positions in the alignment were discarded.
A distance matrix of the remaining 248 sequences was created to check for identical sequences (regarding the 566 positions chosen) deposited with different accession numbers. As a result 58 duplicate sequences were deleted from the alignment, leaving 192 sequences for the phylogeny. Note that similar sequences were not combined to operational taxonomic units (OTUs) in this case. Sequences with a one-nucleotide difference show up as separate entries in the phylogram (but are graphically not resolved in Figure 5).
The phylogeny of these 192 sequences was then calculated for 566 positions of the alignment, beginning downstream of the 63f primer binding site. Due to insertions/deletions, the individual sequences varied from 488 to 543 nt in length - not excluding the 21 nt of the 63f primer. Note that the TReFID analysis was restricted to the 470 nt of a sequence following the 63f motif, but the 566 positions selected for the phylogeny covered the region utilized by TReFID as good as possible. The phylogram was calculated with 1,000 bootstraps resamplings using the Neighbor-Joining algorithm with the Tajima-Nei matrix.
The final phylogram containing 192 sequences is shown in Figure 5. It was drawn as a radial tree (TreeView) and manually edited in Microsoft PowerPoint. It shows three distinct clusters - highlighted by gray ovals - which represent sequences related to either A. brasilense, E. coli, or R. leguminosarum. However, nine of the 192 sequences did not fit in one of these clusters. These were Pseudoalteromonas prydzensis, Pseudoalteromonas sp. Esur-1 , Shewanella gelidimarina, and Idiomarina baltica, as well as sequences from four uncultured proteobacteria (AJ310686 AJ310687 AJ310689 AJ310690). Two of these artifacts are not resolved in Figure 5: Idiomarina sp. LA26 (with I. baltica) and one uncultured proteobacterium.
Sequence homology tables:
| Eco cluster | Rle cluster | Abr cluster | |
| E. coli cluster | --- | 71.1 % | 70.2 % |
| R. leguminosarum cluster | 71.1 % | --- | 80.8 % |
| A. brasilense cluster | 70.2 % | 80.8 % | --- |
As expected the A. brasilense and R. leguminosarum clusters were more closely related (80.8 %) than the other two pairwise combinations. Both strains belong to the Alphaproteobacteria, but to different orders (Rhodospirirllales and Rhizobiales, repectively), whereas E. coli is a Gammaproteobacterium.
|
E. coli (E05133) |
R. leguminosarum (AF533683) |
A. brasilense (AY324110) |
|
| E. coli cluster | 95.2 % | 70.3 % | 70.3 % |
| R. leguminosarum cluster | 70.8 % | 96.0 % | 80.8 % |
| A. brasilense cluster | 70.4 % | 80.7 % | 97.4 % |
The average homology of the E. coli sequence used as input (E05133) to the other 110 sequences within the E. coli cluster was 95.2 %. The respective homologies were 96.0 % for R. leguminosarum related to the R. leguminosarum cluster, and 97.4 % for A. brasilense. The "phylogenetic separation" or "resolution" regarding this control procedure was thus about 5 %. Sequences with pairwise homologies above 95 % were not distinguishable on the basis of their tRF patterns. However, this "resolution" value depends on the sequences picked from the database for such a test and may vary in a small range. Other influential factors are the number of sequences tested and the total size of the respective database.
Matrix: distance matrix of all sequences contained in the TReFID results file version A (as Microsoft Excel file)
Alignment: multiple sequence alignment, basis of the phylogram shown in Figure 5 (in FASTA format)
Phylogram: Neighbor-Joining phylogram calculated with 1000 bootstraps (as phb file, open with, e. g., TreeView)
TReFID results:
(a) for the original SSU
database (version A with 17,462 entries) [TReFID
output] [FASTA
file]
(b)
for the improved SSU database (version B with 21,669 entries) [TReFID
output] [FASTA
file]
Note: The results for (b) contain four more entries due to slight changes in the identification procedure, but are otherwise identical to those of (a). The increased size of the database had no effect in this case as no proteobacterial sequences have been added.